



Materials Studio材料建模與模擬計算工作站方案2021v4 
,計算能帶、態密度√√CASTEP材料科學量子力學程序平面波贗勢密度泛函法,模型較小,包含數十、乃至數百個原子√√NMR CASTEPMS CASTEP的擴展模塊晶胞進行幾何優化,若精度要求較高√ONETEP進行線性標度密度泛函模擬
4106
UltraLAB ??? 4年前
聚合物材料模擬:概述和實例 
1、量子化學 & 密度泛函理論該方法主要針對幾納米或更小的尺度,基于薛定諤方程計算分子和晶體中的電子態。它尤其用于評估材料的電子性質,如激發、極化、分子間力和化學反應。有分子軌道方法(MO)和密度泛函理論(DFT),可以通過將電子密度作為計算目標來減少計算負擔。這些方法中的每一種都根據如何納入電子相關性、如何選擇基函數等進行細分。圖1.
4112 1
上海庭田信息科技有限公司 ??? 3年前
[分析示例] 利用 LDA+U 方法修正半導體帶隙的分析示例 ![[分析示例] 利用 LDA+U 方法修正半導體帶隙的分析示例](https://img.jishulink.com/202404/attachment/54288c5737ea4da892d8ebf41c26bd3c.jpg?image_process=resize,fw_294,fh_172,)
因此,通過模擬使用半導體進行材料設計時,需要準確估算出帶隙,但基于密度泛函理論(DFT)的預測帶隙比實驗結果小得多(圖 1)。 圖 1. 利用 GGA 方法計算的半導體能帶結果(左為硅,右為砷化鎵)用未經校正的 GGA 方法計算出的帶隙大小,硅為 0.72 eV(實驗值為 1.12 eV),砷化鎵為 0.37 eV(實驗值為 1.42 eV)。
2430
上海庭田信息科技有限公司 ??? 2年前
使用Gaussian進行稀土金屬化合物結構優化 
密度泛函理論(DFT)的計算方法,由于其良好的計算效率和準確度,被廣泛應用于稀土化合物的電子結構研究、光學性能、磁性以及催化性能等領域。在這一過程中,Gaussian軟件作為一種經典的量子化學計算程序,提供了豐富的功能來支持DFT計算,廣泛應用于稀土化合物的研究。在稀土化合物的研究中,Gaussian軟件結合密度泛函理論(DFT)能夠在多個方面提供關鍵的理論支持。
2755
320科技工作室 ??? 1年前
Materials Studio中的Castep模塊金屬礦物計算與分析 
金屬礦物屬于大型周期性體系,因此要運用密度泛函理論研究其礦物性質要運用Mterials Studio中的Castep模塊。本文主要講述氧化鋅礦物晶胞模型的構建以及完全解理面的計算和對氧化鋅礦物表面性質分析。
3795
320科技工作室 ??? 3年前
基于MS的Dmol3模塊計算離子液體的電子轉移和成鍵 
然后計算其前線分子軌道、態密度等電子性質。
3193
320科技工作室 ??? 3年前
J-Octa 使用MD和MO/DFT計算相對介電常數 
相對介電常數可以由各個原子電荷偶極矩之和的時間波動得到,公式如下: 分子軌道法計算(MO)/密度泛函理論(DFT)計算MO/DFT讓我們可以估測電子極化,由分子極化率計算相對介電常數。 模擬成果圖2和表1給出了用MO和MD計算苯和丙酮的相對介電常數結果。
3433 11 4
上海庭田信息科技有限公司 ??? 4年前
基于Gaussian高精度熱力學方法計算胺類分子的pKa 
在計算 pKa 值時,Gaussian 軟件通過密度泛函理論(DFT)、Hartree-Fock (HF) 方法、多體擾動理論(MP2)以及更高精度的配置交互(CCSD)等方法進行分子結構優化和能量計算。這些方法能夠提供準確的電子結構信息,為計算 pKa 值提供理論支持。使用量子化學方法精確計算胺類小分子的 pKa 值時,存在一些技術和理論上的難點。
2554
320科技工作室 ??? 1年前
基于VASP的電子結構深度解析 
傳統實驗手段難以直接觀測原子尺度電荷分布與軌道相互作用,而基于密度泛函理論(Density Functional Theory,DFT)的第一性原理計算成為破局關鍵。
3118
320科技工作室 ??? 1年前
近場動力學快速入門程序——板,常規態型本構及兩種求解器(顯示求解和隱式求解) 
第三,彈性材料中定義的應變能密度函數W 是一個將矢量態映射為實數的映射,因此它是一個泛函,而根據frechet導數的定義,?W 是一個將矢量態映射為矢量態的態值函數(即本構模型)。五、本程序包簡介 該文件夾中的算例是一個二維矩形金屬板四邊給定位移的平面應力問題。該算例采用常規態型本構模型以及無網格離散方式,且分別使用顯式求解器和隱式求解器求解。
4038 6 6
FriedPotato ??? 3年前
ISV計劃 | 瀚海量子“雙軟”上架,大幅提升計算體系,共筑國產軟件云仿真生態 | 現招募10名體驗官,軟件免費用 
基于平面波基組的密度泛函理論計算軟件.可快速實現上千原子平面波雜化泛函第一性原理計算核心優勢:1 能計算上萬原子2 在同體系下,計算速度比行業知名軟件快10-30倍3 功能更豐富4 代碼架構新,更新快5 擴展性強第一性原理電子結構計算軟件包,能對周期性復雜大體系的基態電子結構實現線性標度高性能計算核心優勢:1
2255
深圳北鯤云計算有限公司 ??? 3年前
利用機器學習結合實驗揭示非晶氧化鎵原子結構與熱輸運的關系 
近年來,基于密度泛函理論(DFT)或經典力場的分子動力學(MD)模擬一直是建模和理解材料的核心方法。在許多研究中發現,它們的預測能力和可轉移性相對較差。最近,機器學習(ML)技術正在成為一種強大的工具,通過直接從適當選擇的量子力學計算的參考數據集合中映射原子構型和能量之間的關系,有望解決上述材料建模中的挑戰。
2587
熱管理博覽會 ??? 2年前
gaussian-cp2k-lammps-reaxff 專題 
Gromacs對CP2K的結果后處理 專題二 :“Gaussian量子化學計算技術與應用”培訓大綱 課 程 內 容 理論計算化學理論及程序入門操作 1、理論計算化學簡介1.1 理論計算化學概述1.2 HF理論及后HF方法(高精度量化方法)1.3 密度泛函理論和方法
2486 2 2
hdpky ??? 3年前
傳統脆性斷裂相場模型的三維UEL理論及代碼 
然后通過一個與相場相關的裂紋面密度泛函來重構結構內的斷裂能,并將因損傷而退化的變形能與重構的斷裂能代入Francfort-Marigo變分原理就得到了相場模型的基本列式。相場模型中的自變量為兩個連續變化的場,即位移場和相場,因此它可以很方便地由不同數值方法實現。
4489 5 5
dearjj ??? 1年前
Materials Studio零基礎專題培訓重磅來襲 
Amorphous Cell:允許建立無定型系統的代表性模型,并對主要性質進行預測,研究內聚能密度、狀態方程行為等。Reflex:模擬晶體材料的X光、中子以及電子粉末衍射圖譜,幫助確定晶體結構并解析衍射數據。DMol3:獨特的密度泛函量子力學程序,可以模擬氣相、溶液、表面及固體等過程及性質。
2972
320科技工作室 ??? 1年前
計算化學:如何在云平臺上計算聲子譜 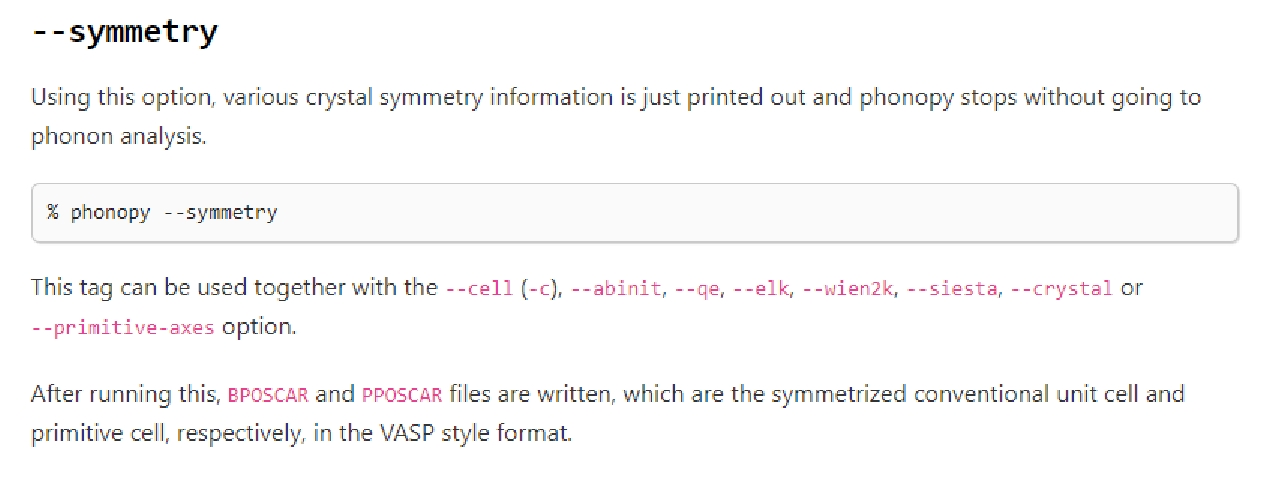
如何擴胞:Phonopy –d –dim=“x x x” –c POSCAR-unitc 擴多大:網傳10A原則 有限差分法/有限位移法/密度泛函微擾理論 后處理:繪制聲子譜圖提取力常數: 命令:phonopy --fc vasprun.xml 調整控制文件 name.conf M_NAME = l Cr DIM =
2633
深圳北鯤云計算有限公司 ??? 3年前
基于CP2K的沸石吸附小分子的結構優化 
由于實驗上難以準確定位骨架 Al 原子,研究者常借助密度泛函理論等計算方法評估不同取代位置和含量對框架穩定性的影響(見 Mater. Today Commun., 26, 102028 (2021))。例如,J. Mater. Chem.
2402
320科技工作室 ??? 9月前
VASP結合vaspkit+ShengBTE計算熱電優值(一) 
前者可用于DFPT(密度泛函微擾)方法,后者應用于有限位移法。兩種方法計算的結果沒有區別。 (3)DFPT法DFPT法需要使用SPOSCAR進行計算(單個任務)。可參考王寧博士在B站的視頻[7]。筆者在這里貼出自己的代碼僅供參考。
2891
320科技工作室 ??? 2年前
面向金屬增材制造的拓撲優化設計研究進展 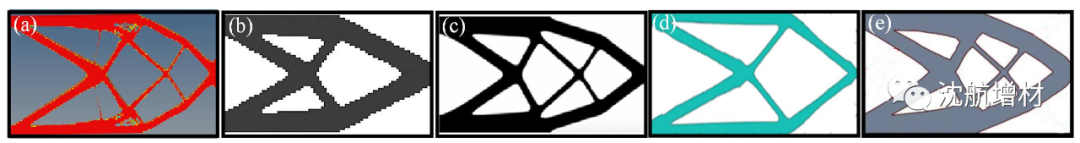
Chen 等 在目標函數中引入二次能量泛函,將幾何特征尺寸信息引入水平集框架,實現梁狀柔性機構最小尺寸優化。隨后 Luo 等 將二次能量泛函引入無鉸鏈柔性機構,對原始目標函數進行增廣,采用半隱式算法,避免傳統水平集方法存在的數值求解困難問題,實現更為高效的結構最小特征尺寸控制與優化,但該方法未提供明確的幾何信息,無法實現結構最小尺寸的精確控制。
4053
張偉一 ??? 2年前
劉名瑞 等:基于物理吸附儲氫材料的研究進展 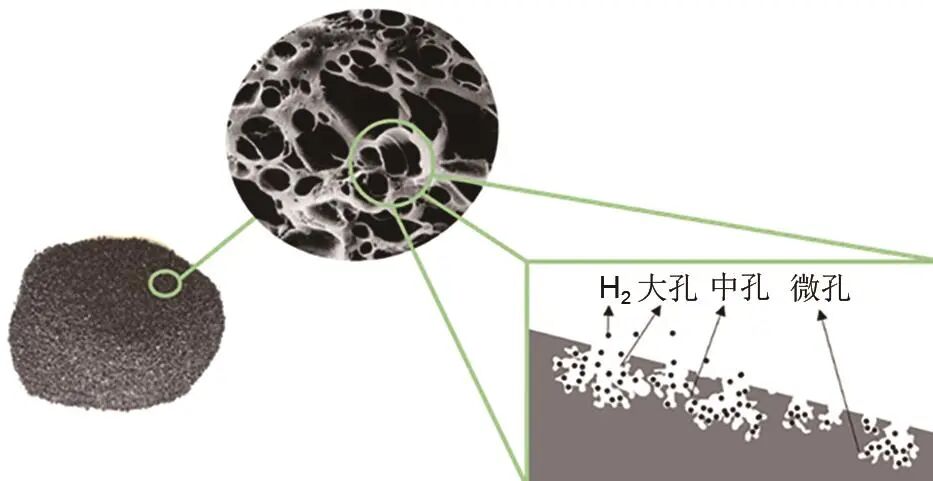
Bi等設計了一種新型碳基富勒烯結交叉納米管周期網絡結構,通過密度泛函理論計算和分子模擬,得到Li@C 158B 22、Na@C 132B 24和Be@C 132B 24體系,分別可以吸收13.95%、10.09%和10.85%的氫氣。
6268 3 2
能源阿陽 ??? 2年前
20條/頁 
跳至頁

技術鄰APP
工程師必備
工程師必備
- 項目客服
- 培訓客服
- 平臺客服
TOP
