藥物設計中的分子動力學
分子動力學(MD)模擬是基于結構的藥物設計的有用工具。一些情況下,在對接之前已經進行了蛋白質靶標的MD模擬以產生與晶體結構不同的可用蛋白質構象異構體。此外,在對接后進行MD模擬,評估命中化合物的預測結合模式作為最終在計算中和引導化學合成用于命中優化的過濾器。
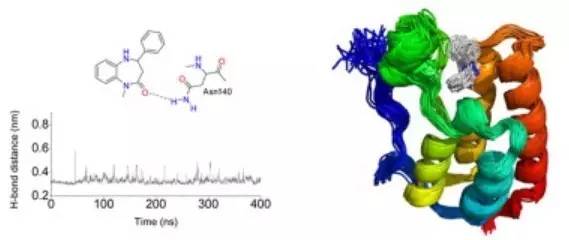

1.MD作為分子片段映射到結合位點的工具
研究人員提出了MD作為工具分析蛋白質與小分子的結合自由能表面和結合途徑。由于可用的晶體結構和結合親和力的測量,研究人員將MD應用于FKBP的肽基脯氨酰順反異構酶和六個配體之間的非氫原子。研究人員通過色氨酸熒光猝滅測定法測得FKBP的親和力在高μM至低mM范圍內。對于每個配體產生了結合過程的構象空間網絡。
第一步中,沿著多個軌跡保存的相對位置和方向根據一組分子間距離進行聚類。集群被認為是網絡的節點,并且在MD模擬期間觀察到這些集群之間的直接轉換是網絡的鏈路。有趣的是,網絡分析揭示了以不同的分子間氫鍵和疏水接觸為特征的多種結合模式。此外,解離動力學顯示單指數時間依賴性,這表明完全解離的屏障顯著高于不同結合模式之間的屏障。
比較實驗和模擬方法是有啟發性的,用于分析片段與蛋白質結合的上述生物物理技術在時間和空間分辨率方面具有局限性。相比之下,MD模擬在原子細節水平上產生了自由能表面和結合途徑的完整圖像。
2.通過MD選擇構象異構體
蛋白質靶標的晶體結構表示折疊狀態下的許多狀態之一。大多數情況下,折疊狀態的整體拓撲在其子狀態中是保守的,但是在結合位點中甚至單個側鏈的不同方向可以顯著影響對接結果。
McCammon小組開發了一種方案來解決對接中的蛋白質柔性:首先,對載脂蛋白的MD模擬進行廣泛構象空間采樣。第二階段涉及到一個大型MD系綜幀的對接。
在方案的初始之后的組合對接發現了幾種潛在的抑制劑,這不可能通過對接原始的晶體結構獲得。
案例
為了選擇用于高通量對接的西尼羅病毒非結構3蛋白酶(NS3pro)的結構,將三個分子片段(苯、甲基胍和2-苯基咪唑啉)放置在100個幀中,運行MD模擬。使用苯,因為它是小分子藥物中最常見的環,且NS3pro活性位點的表面具有負靜電勢;基于已知NS3pro抑制劑的信息,故使用其他兩個帶有正電荷的片段。選擇對三個片段都非常有利的碎片多樣性集合對接到MD幀中。經過過濾和目視檢查后,在體外僅測試了5個分子,其中2個顯示出低微摩爾的親和力。重要的是兩種活性化合物可以對接到晶體結構中,但是它們沒有通過過濾器,換句話說,它們在MD幀的結構中是陽性的,在晶體結構中是陰性的。

3.通過MD誘導契合(MD-IF)準備結合位點
研究人員設計了MD協議采樣與晶體結構不同的蛋白質構象。這些構象揭示了天然配體或底物的結合位點中活性位點側鏈或一些口袋中較大孔的側鏈取向。另外,通過已知抑制劑的信息,使用來自載脂蛋白靶標的MD模擬的結構整體不會偏向于可能導致完全新穎的抑制劑。
案例
蛋白激酶中的ATP結合位點位于催化結構域的兩個葉片之間,特別是在活化環、富含Gly環和一些側鏈。小分子抑制劑可以促進環和側鏈的置換,這種重排效應形成的口袋稱為變構位點,其接近ATP結合位點,且在許多有效的選擇性酪氨酸激酶抑制劑中被疏水部分占據。最近的一項研究中,通過MD產生了酪氨酸激酶EphA3活性位點的誘導擬合,以適應不能停靠在EphA3晶體結構中的已知II型抑制劑。MD誘導擬合(MD-IF)結構的MD幀用于大約175,000個化合物藥效團定制的化合物庫的高通量對接,其在體外測試的12個分子中產生3個活性物質,命中率為25 %。重要的是EphA3激酶的ATP結合位點中環的誘導位移和Tyr742側鏈的重新取向不能通過常規的能量最小化來實現。

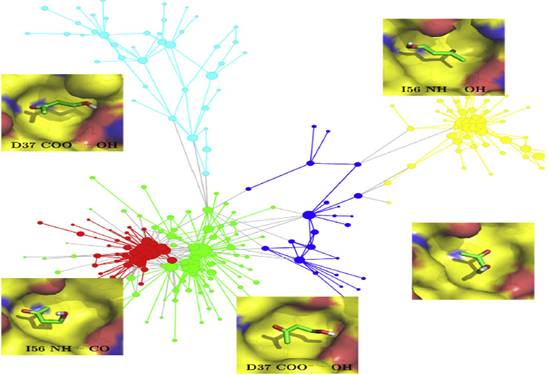
4.預測結合模式的MD驗證
有兩種類型的MD用于驗證通過對接獲得的結合模式:
第一種,在高吞吐量對接和排名之后,使用MD模擬作為最終的過濾器。這種方法本質上是平行的,因為對于一組候選配體運行獨立的MD模擬,并且可以并行地分析軌跡。
第二種使用活性化合物的MD模擬作為指導化學合成以進行命中化合物或者先導化合物的優化。當復合物晶體結構不可用時,MD采樣獲得的原子信息特別有用。
案例
研究人員進行了基于片段的高通量對接篩選,以發現與BRD4第一個溴結構域中乙酰賴氨酸結合位點競爭的小分子。從665,184個市售分子的化合物庫開始,近5000種化合物在預測結合能和配體效率的過濾器中保留下來。依據命中化合物的姿勢,將55個具有不同錨定片段的化合物進行多次MD模擬進入BRD4的第一個溴結構域,以評估主要的結合相互作用。MD模擬顯示,一些選定化合物的結合模式不穩定,且該信息用于減少體外驗證的化合物的數量。最后,僅購買了24種化合物,其中4種化合物在單位數量級范圍內表現出親和力。研究人員已解析測試抑制劑與BRD4第一個溴結構域的復合物晶體結構,其中兩種抑制劑,通過對接和MD預測的結合模式與晶體結構中的結合模式相同。

5.MD揭示已知藥物和天然配體的結合模式
研究人員已經采用MD模擬來再現在晶體結構中觀察到的配體-蛋白質結合模式,以及使用MD來闡明其結合模式未知的藥物作用機制。此外,MD模擬也用來揭示天然配體到表觀遺傳靶的結合模式。
案例:JAK2激酶抑制劑ruxolitinib和SAR302503的結合模式
激酶抑制劑結合模式的原子水平信息非常有助于解釋其作用機制、分析其特異性和可能的抵抗機制。此外,還可用于優化化學衍生物以提高效力和特異性。Ruxolitinib和SAR302503是用于治療骨髓惡性腫瘤酪氨酸激酶JAK2的兩種強效抑制劑。然而,他們對JAK2的結合模式尚未有報道。最近,研究人員通過多次MD模擬確定了這兩種抗癌藥物的結合模式,抑制劑以其活性構象靶向JAK2的ATP結合位點。MD模擬結果表明,Ruxolitinib的雙環支架與JAK2的鉸鏈區域有兩個持久的氫鍵。MD模擬預測的SAR302503的結合模式由鉸鏈區域中的氨基嘧啶支架和Leu932的骨架極性基團之間的兩個氫鍵穩定。通過MD模擬獲得的結合模式在原子細節上解釋了體外觀察到的抗性引起點突變的作用。此外,它們作為用于解釋預期在用這兩種JAK2抑制劑治療的患者中出現的突變的模板非常有用。

總結
藥物相關蛋白質靶標在其載脂蛋白狀態與配體復合物的全原子MD模擬在高通量對接的開始和最后階段提供了有用的信息。載脂蛋白結構的顯式溶劑MD模擬正在變得越來越頻繁地選擇一個或多個幀用于對接大型化合物庫。在排名的最后階段,從預測結合模式開始的MD模擬用于對命中化合物的計算驗證。
此外,原子MD模擬已經提供了對抗病度藥物地瑞納韋機制的理解;有助于解釋在臨床中使用的兩種激酶抑制劑作為抗癌藥物的選擇性;并且已經揭示了乙酰賴氨酸具有比在可用的晶體結構中觀察到的更多的替代結合模式。所有例子表明,MD的應用將在未來的藥物設計中發揮更重要的作用。通過高通量對接鑒定有效抑制劑有三個主要障礙:化合物庫非常小的化學空間、用于評分的近似值和使用剛性蛋白質結構。本文中綜述的MD方案和未來基于MD計算篩選方法的發展將有助于消除后兩種障礙。

參考資料:
1.Zhao H, Caflisch A. Molecular dynamics in drug design[J]. European journal of medicinal chemistry, 2015, 91: 4-14.
2.Siu M, Pastor R, Liu W, et al. 2-Amino-[1, 2, 4] triazolo [1, 5-a] pyridines as JAK2 inhibitors[J]. Bioorganic & medicinal chemistry letters, 2013, 23(17): 5014-5021.
3.Zhou T, Georgeon S, Moser R, et al. Specificity and mechanism-of-action of the JAK2 tyrosine kinase inhibitors ruxolitinib and SAR302503 (TG101348)[J]. Leukemia, 2014, 28(2): 404.





















工程師必備
- 項目客服
- 培訓客服
- 平臺客服
TOP




