基于Gromacs的蛋白分子動(dòng)力學(xué)模擬(RMSD、RMSF及蛋白的回旋半徑)

一、實(shí)驗(yàn)要求
實(shí)驗(yàn)對(duì)象:目標(biāo)體系為modeller或其他方法建模的結(jié)果中評(píng)價(jià)最好的模型。
軟件: Gromacs-5.1.2
二、實(shí)驗(yàn)步驟
加立場(chǎng)
gmx pdb2gmx –h 打開幫助菜單。 選力場(chǎng)的時(shí)候選擇 Amber99sb…,溶劑類型選Tip3p。
2、加模擬盒子,溶劑層厚度為0.8nm。
gmx editconf -bt ( boxtype: 做三個(gè)盒子的對(duì)比cubic/triclinic/dodecahedron ), -d 0.8 , 比較三種類型的盒子水分子數(shù)目差別。
3、加水溶劑。
gmx solvate –h
4、做能量?jī)?yōu)化。 參數(shù)文件:em.mdp(可從官網(wǎng)上下載) etol: 500,達(dá)到收斂,該步驟完成。
5、平衡體系,將體系升溫。 從0K升溫到300K,在30ps內(nèi)完成。
6、動(dòng)力學(xué)模擬采樣。模擬時(shí)間:1ns,步長(zhǎng):2fs。坐標(biāo)保存的頻率為每10ps保存一幀結(jié)果,整個(gè)軌跡共100個(gè)frame.
7、結(jié)果分析:
7.1. 全體系的alpha-C原子的均方根偏差(RMSD)結(jié)果獲取及分析,gmx rms 。
7.2. 全體系的alpha-C原子的均方根漲落(RMSF)結(jié)果獲取及分析, gmx rmsf。
7.3. 體系的總勢(shì)能變化曲線分析,采用g_energy命令。
7.4. 分析蛋白質(zhì)的回旋半徑變化,采用g_gyrate命令
7.5. 將采樣最后的構(gòu)象與初始構(gòu)象進(jìn)行疊加比較,分析構(gòu)象的變化情況。
7.7. 從模擬的軌跡中將體系中的蛋白質(zhì)單獨(dú)取出來,另存為一個(gè)軌跡文件protein.xtc, 用VMD的插件”movie maker”做成一個(gè)小電影。
三、操作過程記錄及結(jié)果
Step1: 輸入如下命令,在命令行的交互式操作中,選擇力場(chǎng)(輸入5)AMBER99SB protein,選擇溶劑模型(輸入1):TIP3P
gmx pdb2gmx -f CCM.B99990002.pdb -o conf.gro -p topol.top
選擇力場(chǎng)和溶劑模型后出現(xiàn)如下屏幕回顯(部分):
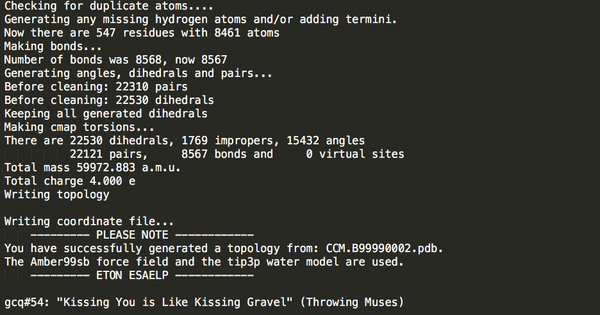
Step2:加盒子。輸入如下命令:分別加三個(gè)模擬盒子,比較三種類型的盒子水分子數(shù)目差別
gmx editconf -f conf.gro -bt cubic -d 1.0 -o cubic_out.gro
gmx editconf -f conf.gro -bt triclinic -d 1.0 -o triclinic_out.gro
gmx editconf -f conf.gro -bt dodecahedron -d 1.0 -o dodecahedron_out.gro
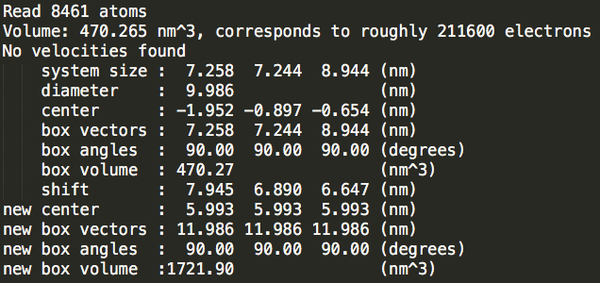
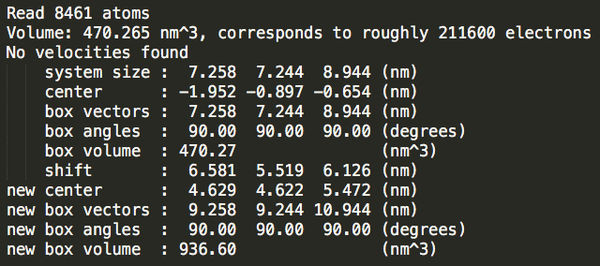
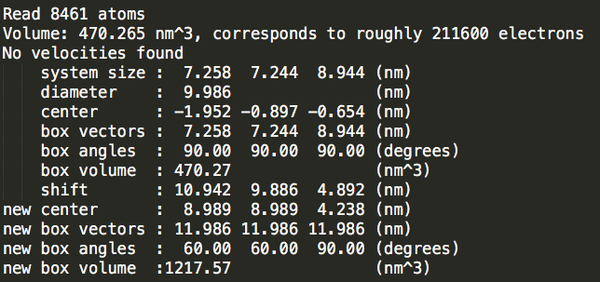
Step3:加溶劑。
gmx solvate -cp cubic_out.gro -o cubic.pdb -p topol.top
gmx solvate -cp triclinic_out.gro -o triclinic.pdb -p topol.top
gmx solvate -cp dodecahedron_out.gro -o dodecahedron.pdb -p topol.top
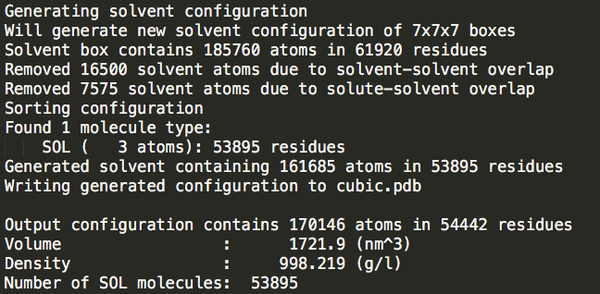
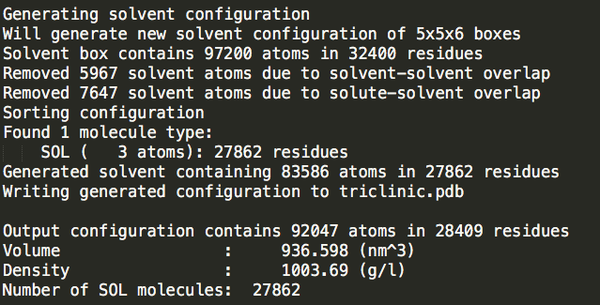
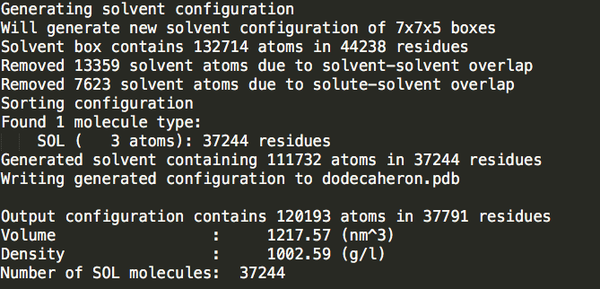
cubic_out.gro
triclinic_out.gro
dodecahedron_out.gro
53895
27862
37244
可以發(fā)現(xiàn)立方體盒子里水分最多。
Step4:做能量?jī)?yōu)化, etol: 500,達(dá)到收斂,該步驟完成。
從官網(wǎng)上下載一個(gè)em.mdp的例子文件,下載地址如下
http://www.bevanlab.biochem.vt.edu/Pages/Personal/justin/gmx-tutorials/complex/Files/em.mdp 檢測(cè)離子平衡
輸入如下命令,檢測(cè)體系的離子平衡:
gmx grompp -f em.mdp -c cubic.pdb -p topol.top -o cubic_em
檢測(cè)結(jié)果如下:發(fā)現(xiàn)需要添加四個(gè)負(fù)離子
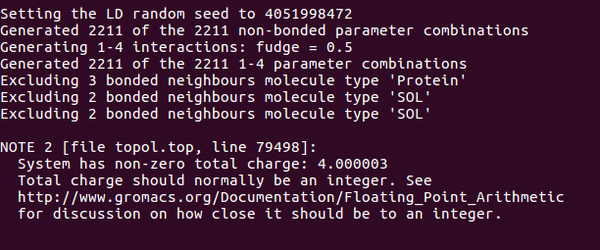
Step5:添加離子,發(fā)現(xiàn)總共是4個(gè)正電荷,需要加4負(fù)電荷 添加離子命令:
gmx genion -s cubic_em.tpr -o ION.gro -p topol.top -nn 4 -pname NA 選擇(SOL)
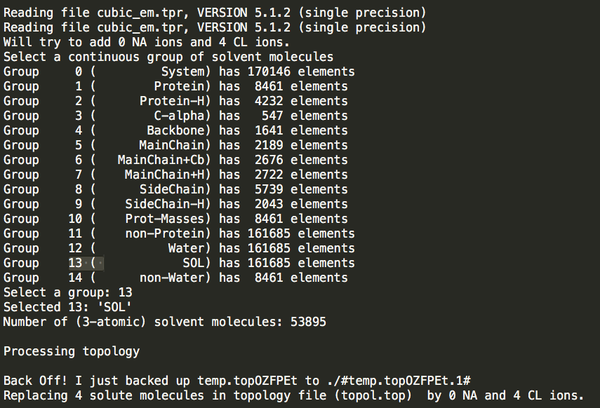
加離子后:
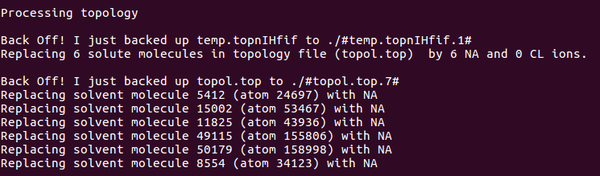
Step6:再次檢測(cè)離子平衡,使用新的文件ION.gro進(jìn)行檢測(cè):
gmx grompp -f em.mdp -c ION.gro -p topol.top -o cubic_em.tpr
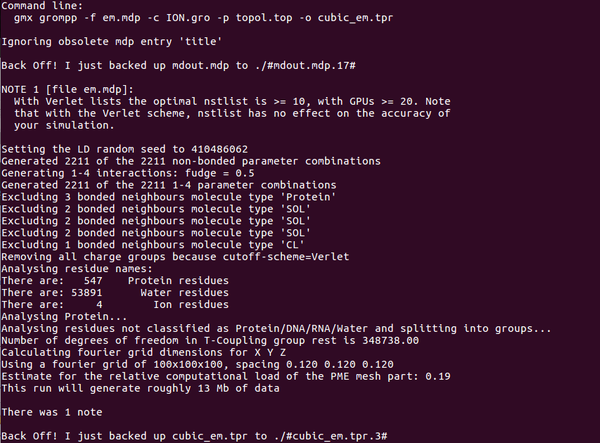
能量最小化:
gmx mdrun -s cubic_em.tpr -deffnm cold -v
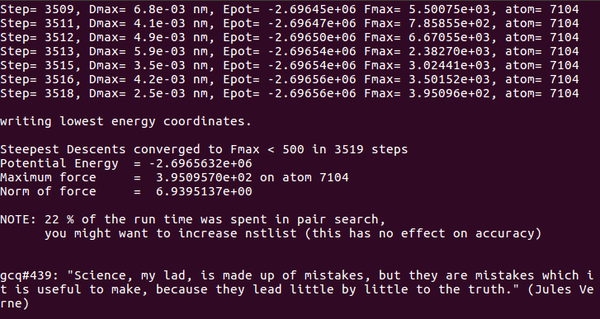
能量查看(potential)
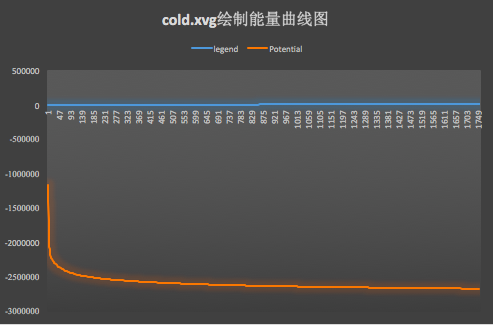
gmx energy -f cold.edr -o cold.xvg
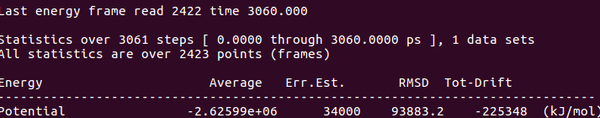
將cold.xvg導(dǎo)入Excel繪制能量曲線圖,看能量是否降低,如下圖:
可以發(fā)現(xiàn)能量確實(shí)降低了。
Step7:平衡體系,將體系升溫。 從0K升溫到300K,在30ps內(nèi)完成
根據(jù)生成的mdout.mdp文件,創(chuàng)建uprade.mdp,將體系升溫至300k,步長(zhǎng)為15000,改步長(zhǎng):

gmx grompp -f upgrade.mdp -c cold.gro -p topol.top -o cubic_em_hot.tpr 回顯如下:
gmx mdrun -s cubic_em_hot.tpr -v -deffnm hot 回顯如下:
然后進(jìn)行能量查看(選擇tempratrue):
gmx energy -f hot.edr -o hot.xvg 回顯如下:
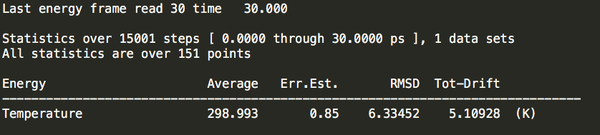
將hot.xvg導(dǎo)入excel表格發(fā)現(xiàn)確實(shí)升到了300k
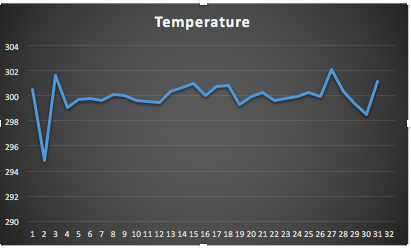
Step6: 體系采樣
gmx grompp -f sample.mdp -c hot.gro -p topol.top -o cubic_em_hot_sample.tpr
sample.mdp需要根據(jù)老師要求進(jìn)行修改,修改地方如下:
模擬時(shí)間:1ns,步長(zhǎng):2fs。坐標(biāo)保存的頻率為每10ps保存一幀結(jié)果,整個(gè)軌跡共100個(gè)frame
輸入命令:gmx mdrun -s cubic_em_hot_sample.tpr -v -deffnm hot_sample
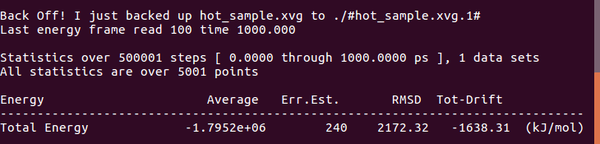
能量查看(temperature),能量確實(shí)達(dá)到了300K(約等于)
gmx energy -f hot_sample.edr -o hot_sample.xvg
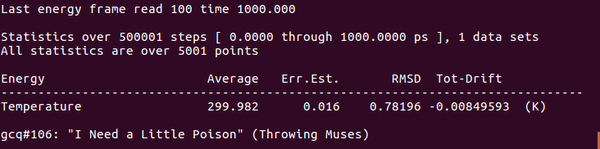
Step7:結(jié)果分析與討論
1、全體系的alpha-C原子的均方根偏差(RMSD)結(jié)果獲取及分析
輸入如下命令:gmx rms -s cubic_em_hot_sample.tpr -f hot_sample.trr -o rmsd.xvg
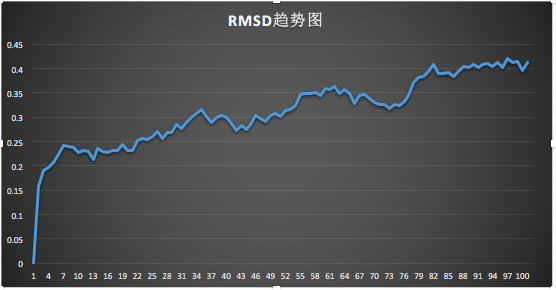
RMSD值可以反應(yīng)出模擬過程中體系的穩(wěn)定情況,將rmsd.xvg導(dǎo)入EXCEL表,繪折線圖,由圖可知?jiǎng)恿W(xué)模擬逐漸達(dá)到平衡。
全體系的alpha-C原子的均方根漲落(RMSF)結(jié)果獲取及分析
輸入如下命令:gmx rmsf -s cubic_em_hot_sample.tpr -f hot.trr -o rmsf.xvg
RMSF計(jì)算每個(gè)原子相對(duì)于其平均位置的漲落, 表征了結(jié)構(gòu)的變化對(duì)時(shí)間的平均, 給出了蛋白各個(gè)區(qū)域柔性的表征, 對(duì)應(yīng)于晶體學(xué)中的b因子(溫度因子). 通常, 我們預(yù)期RMSF和溫度因子類似, 這可以用于考察模擬結(jié)果是否與晶體結(jié)構(gòu)符合。
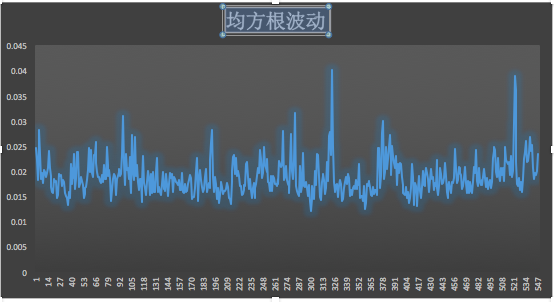
3、體系的總勢(shì)能變化曲線分析
gmx energy -f hot_sample.edr -o potential.xvg
可以看出總勢(shì)能總體在波動(dòng) :
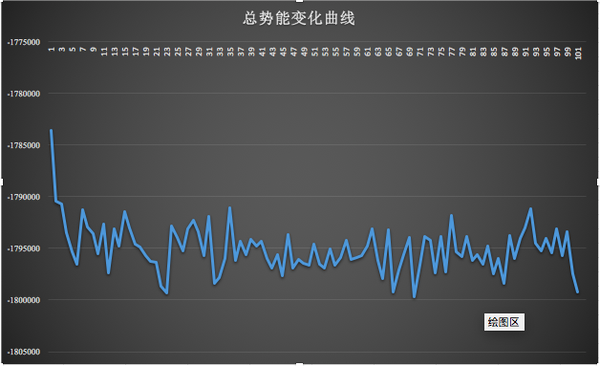
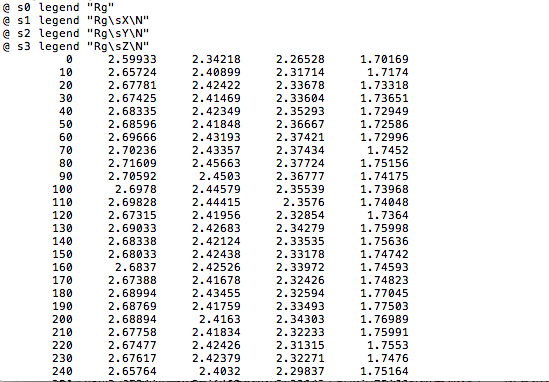
4、分析蛋白質(zhì)的回旋半徑變化(只繪制了半徑的曲線圖)
gmx gyrate -f hot_sample.trr -s cubic_em_hot_sample.tpr -o gyrate.xvg
回旋半徑是描述蛋白質(zhì)緊密型的一個(gè)物理量,半徑越小說明致密性越好,,即蛋白質(zhì)結(jié)構(gòu)就越穩(wěn)定。
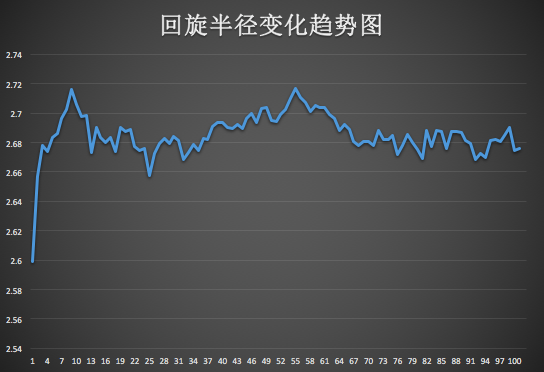
分析蛋白質(zhì)的回旋半徑變化(只繪制了半徑的曲線圖),如圖第一列是蛋白質(zhì)的回旋半徑:
由圖可看出蛋白質(zhì)的回旋半徑有逐漸變小。
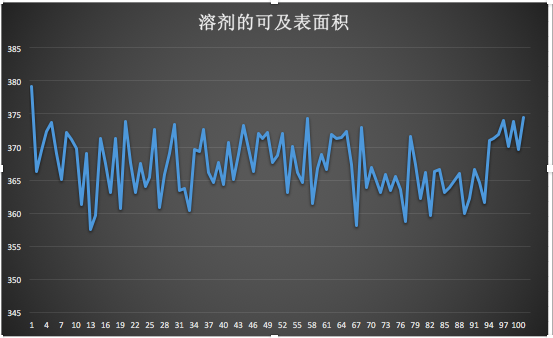
5、分析溶劑的可及表面積(SASA)選擇SOL
gmx sasa -f hot_sample.trr -s cubic_em_hot_sample.tpr -o area.xvg
它是描述蛋白質(zhì)疏水性的重要手段,氨基酸殘基的疏水性是影響蛋白質(zhì)折疊的重要物理作用
6、將采樣最后的構(gòu)象與初始構(gòu)象進(jìn)行疊加比較,分析構(gòu)象的變化情況
gmx confrms -f1 ION.gro -f2 hot_sample.gro -o fit.pdb
感覺用肉眼看不太出來,變化不太大。
Step8:VMD小電影制作
用最新生成的liuwanlin.pdb做vmd小電影。
導(dǎo)入pdb——選擇Extension->visualization->movie maker,Movie duration(seconds)設(shè)置生成視頻的總時(shí)長(zhǎng),設(shè)定20s,使用錄屏軟件錄小視頻。
最后,有需要?dú)g迎通過微信公眾號(hào)聯(lián)系我們。
微信公眾號(hào):320科技工作室。





















工程師必備
- 項(xiàng)目客服
- 培訓(xùn)客服
- 平臺(tái)客服
TOP




